Passion, inspiration, perseverance, altruism, and need have historically been the factors driving innovators. During World War II, antibiotics were scarce and chronic osteomyelitis and bone deficiencies such as non-unions and deformities were common. G.A. Ilizarov, a general surgeon in the Ural Mountains in Siberia at the time, saw a majority of his patients suffering from musculoskeletal injuries. He used modular ring external fixators with tensioned transosseous wires to stabilize bone fragments, introducing the concept of “local bone regeneration” using minimally invasive surgery. His method salvaged limbs, allowing disabled soldiers to avoid painful amputations and return to normal activity. While Ilizarov’s innovation was a result of necessity, it also stemmed from a passion to improve treatment for his patients. Today, large-scale innovation is scarce, not due to the lack of ideas, but rather to the difficulty of executing and implementing those notions. The biggest hurdle in getting a medical device to market is a lack of understanding of governmental regulatory processes. This has become an important part of a product’s lifecycle when transitioning from concept to implementation, particularly in the field of orthopedic products. This column discusses the current U.S. regulatory system for medical devices and its effects on innovation in the implantable orthopedic device industry.
Medical Device Regulatory Pathways
The pathway to market for new medical devices depends on the level of risk associated with that device. Higher risk devices, as defined by the U.S. Food and Drug Administration (FDA), must meet particularly stringent information criteria to be marketed in the United States (Table 1).
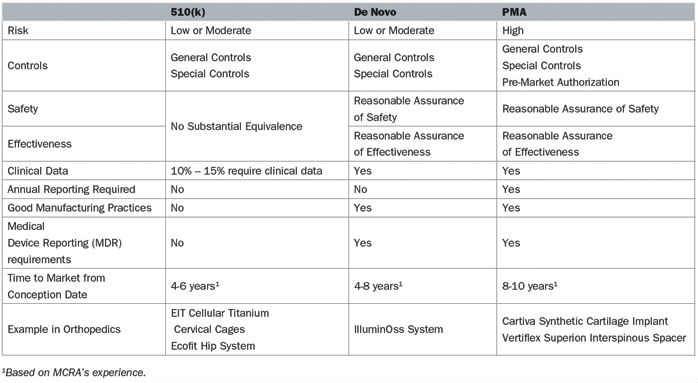
Table 1: Comparison of the De Novo, 510(k), and PMA pathways in 2018
A 510(k) is the most common regulatory pathway utilized by manufacturers, is appropriate for low- or moderate-risk devices, and is not often correlated with innovation. These devices attain marketing clearance by determining substantial equivalence to a predicate, specifically a product currently on the market. Substantial equivalence is defined as similarity of the devices with respect to intended use, design, energy used or delivered, materials, chemical composition, manufacturing process, performance, safety, effectiveness, labeling, biocompatibility, standards, and other characteristics, as applicable.
In many cases, the most innovative devices are deemed high-risk due the absence of comparable products and follow either the de novo or PMA pathway. Truly high-risk devices, in which insufficient information exists to determine whether general and special controls are sufficient to provide reasonable assurance of the product’s safety and effectiveness, follow the PMA pathway. Novel devices that do not have a predicate are classified in the highest risk class, regardless of the level of actual risk it poses or the ability of general and special controls to assure safety and effectiveness. The de novo process allows these novel devices with low to moderate risk to be reclassified from a high-risk class, which requires a PMA, to a low or moderate risk class, which only entails a 510(k).
Medical Device Innovation and the FDA
Despite boasting the world’s largest medtech market and laying claim to some of the industry’s largest corporations the development of innovative devices is lagging in the United States.2 Medtech innovation is a broad term that can encompass any individual part of the product lifecycle such as design, function, or manufacturing. Product innovation can refer to the invention of new devices or the improvement of existing ones and can be either radical or incremental. Radical innovation is characterized by dramatic change while incremental innovation is defined by small changes that may or may not eventually lead to radical ingenuity. Innovation can also include adoption—taking an existing device and applying it in a way it was not previously intended, which often occurs in cross-industry innovation. According to an analysis of FDA databases from 2015 to 2017, 97.9 percent of submissions were 510(k)s while only 0.8 percent and 1.4 percent were de novos and PMAs, respectively. In the orthopedic industry specifically, 99.4 percent were 510(k)s and 0.6 percent were PMAs (Figure 1).
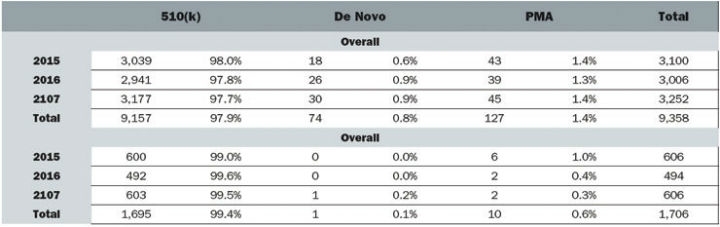
Figure 1: Medical Device Submissions (2015-2017)
While the U.S. orthopedic device sector remains strong (estimated to surpass $25 billion in value by 2024), these numbers show a disparity of innovative products coming to market.1,3 Aligning with the trend towards preventing and curing disease, the orthopedic industry is likely to see growth from complex products such as instrumentation for minimally invasive surgery, combination devices, and customization in the near future.
Impact of Regulation on Innovation
The U.S. regulatory environment for medical devices generally inhibits the development of truly innovative products, as evidenced by the trend toward a greater number of submissions for incremental or low- to moderate-risk technologies. The current regulation for assessing the safety and efficacy of medical devices was developed in 1976; the scope and types of devices currently in development are quite different than those envisioned 40 years ago. Consequently, the industry is questioning whether the regulations are still ideal for ensuring product safety and effectiveness without adding undue burden to the manufacturer. According to MCRA’s review of FDA’s published guidance documents, there currently are 874 guidance documents specific to medical devices alone, compared with 318 in 1999. In addition, increasing non-clinical and clinical data requirements outlined in these guidance documents add cost and time in order to bring these devices to the market. This increasingly complex regulatory system leads to medical device companies having difficulty navigating the pathway towards bringing their latest inventions to customers.
The result of this increased regulatory burden corresponds to a substantial increase in the cost of bringing a device to market. In 2014, this cost under the 510(k) pathway was about $31 million, compared to about $94 million under the PMA pathway.4 In order to promote innovation in the United States, steps must be taken to correct the shortcomings of the regulatory process and create a landscape where developing innovative technologies is in the best interest of the industry.
Conclusions
The current industry landscape for innovative medical devices has become more competitive and challenging, with significant regulatory hurdles blocking the path to the U.S. market. While the FDA’s first priority should always be to ensure public safety, the agency also has an important responsibility to provide the U.S. population with access to innovative and beneficial products. The Center for Devices and Radiological Health at the FDA recently released a 2018-2020 Strategic Priorities Document outlining its commitment to better public access to high-quality innovative technology, outlining a two-year goal to help at least 50 percent of medtech manufacturers debut their devices first in the United States or in parallel with other major markets (the deadline is Dec. 31, 2020).5 In light of this announcement, FDA reviewers should continue focusing on transparency and providing clear guidance to industry while also using their scientific expertise to exercise flexibility to relieve the burden of the regulatory process whenever possible. Simultaneously, medical device companies should consider the regulatory process from the beginning of the product’s lifecycle and communicate openly with the FDA to ensure efficiency. Nonetheless, healthcare is entering an exciting period and medical devices have the opportunity to make an enormous impact on prevention, treatment, and eradication of disease. Proper collaboration between companies and regulatory bodies will accelerate the rate at which innovative, game-changing technologies are available to the public.
References